
Introduction
Neurological combination products—integrating drugs, devices, and/or biologics—are transforming treatment for central and peripheral nervous system disorders. As conditions like epilepsy, Parkinson’s disease, multiple sclerosis, and chronic pain demand more targeted approaches, the convergence of pharmaceuticals and medical technologies offers promising solutions. These hybrid therapies enhance delivery, address complex disease mechanisms, and improve outcomes. They are advancing neurological care and expanding clinical possibilities.
Yet, this innovation introduces significant regulatory challenges. The U.S. Food and Drug Administration (FDA) considers neurological combination products among the most difficult to evaluate due to the complexity of the nervous system, the integration of technologies, and their heightened risk profile.
The Evolving Landscape
Combination products are defined as therapeutic and diagnostic products that combine drugs, devices, and/or biological products. These products can take various forms:
- Drug-device: For example, drug-eluting stents or inhalers delivering bronchodilators
- Biologic-device: Such as scaffold-based tissue regeneration platforms using growth factors
- Drug-biologic: Like antibody-drug conjugates used in targeted therapies
In the neurological field, this convergence is especially vital. As neurological disorders often involve complex and chronic conditions, a single-modality treatment may fall short. Combining drugs with precision-targeted devices can facilitate localized delivery, sustained release, and adaptive response to real-time neural feedback. Notably, the rise in neurodegenerative diseases due to aging populations has further fuelled innovation in this domain. The industry is witnessing an explosion in brain-targeting implants, wearable neurotech, and advanced drug delivery systems—each blurring traditional therapeutic boundaries.
Real-world Example:
Intrathecal pumps: Intrathecal pumps are reshaping neuromodulation by enabling the direct infusion of potent drugs into the cerebrospinal fluid, effectively bypassing systemic circulation and the restrictive blood-brain barrier. This localized delivery achieves therapeutic concentrations at the spinal cord or brain with unmatched precision, levels that systemic administration often cannot reach. The result is a significantly enhanced clinical effect using lower drug doses, with reduced systemic toxicity and side effects.
FDA Framework and Evaluation Pathways
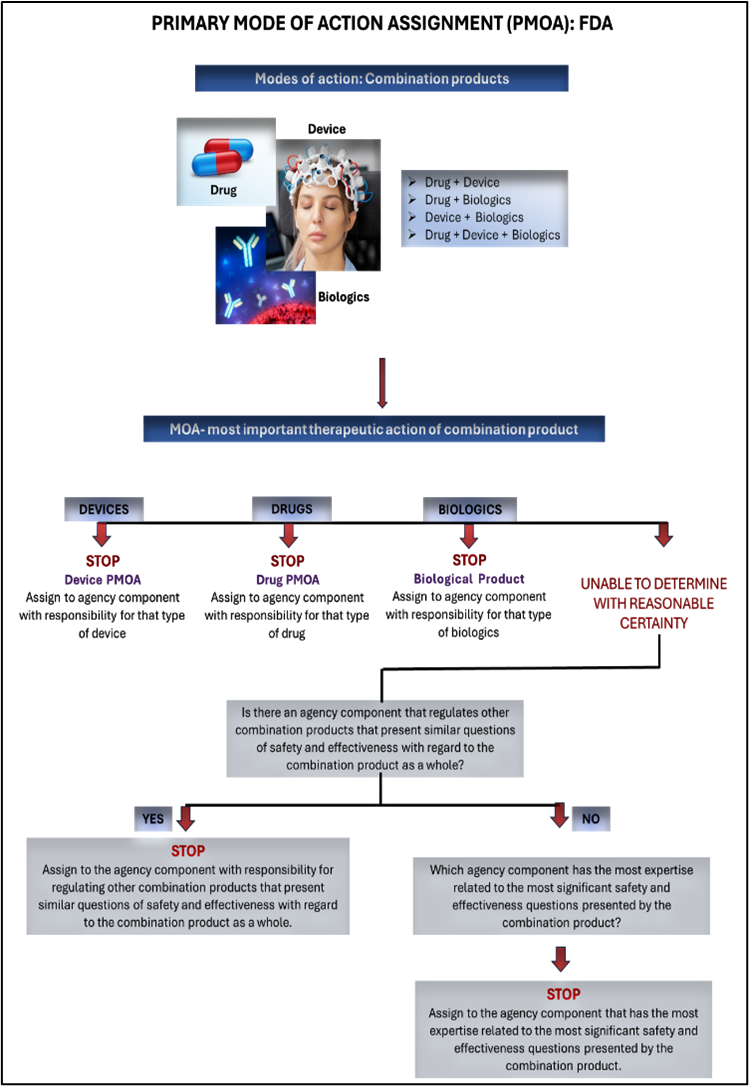
Combination products are regulated by the FDA’s Office of Combination Products (OCP), which determines the primary mode of action (PMOA). The PMOA dictates which FDA centre takes the lead in reviewing the application: CDER (Center for Drug Evaluation and Research), CDRH (Center for Devices and Radiological Health), or CBER (Center for Biologics Evaluation and Research).


{kind=link}