FDA Inspection Observations & Form 483: Practical Insights for Industry, Clinical & Regulatory Professionals
August 6, 2025

As FDA inspection activities intensify post-pandemic, FDA Form 483 (also known as Inspectional Observations) has regained prominence as a critical regulatory checkpoint. Inconsistent data integrity, incomplete documentation, and procedural gaps remain among the top-cited issues. For pharmaceutical, biologics, and device companies alike, a single observation can have serious downstream consequences.
This issue unpacks the essentials of Form FDA 483—its purpose, implications, and best practices for response—while offering industry-focused strategies to manage inspection outcomes with confidence.
What is FDA Form 483?
FDA Form 483, officially titled “Inspectional Observations,” is issued by the U.S. Food and Drug Administration’s (FDA) investigators after an inspection when they observe any conditions that may constitute violations of the Federal Food, Drug, and Cosmetic Act (FD&C Act) and related laws.
- Purpose: To notify company management of important concerns about facility conditions, processes, recordkeeping, personnel practices, or data integrity.
- Scope: Applies to drugs, biologics, medical devices, dietary supplements, and more.
- Timing: At the end of an inspection (routine, for-cause, or pre-approval).
- Not a Final Decision: A Form 483 is not a final agency decision or enforcement action—but it is often a precursor to one.
Regulatory Significance: What’s at Stake?
Legal and Compliance Implications
Although not a formal enforcement action, receipt of a FDA Form 483 can result in:
- ❗ Warning Letters
- 🚫 Import Alerts
- 📉 Product Recalls
- 🛑 Approval Withholdings (e.g., NDA/BLA)
- 📝 Consent Decrees
Company Response
- Firms are strongly encouraged to respond in writing within 15 business days.
- A well-documented corrective and preventive action (CAPA) plan can mitigate regulatory escalation.
Where to Access Previous FDA 483 Observations?
FDA maintains several databases for inspection transparency:
- 📊 FDA Inspection Observations Database – Viewable by fiscal year in downloadable Excel.
- 📌 Inspection Classification Database
- ⚠️ FDA Warning Letters
- 🗃️ Frequently Asked Questions about Form 483
These resources help identify trends, benchmark issues, and guide preparation.
💡 TIP: Monitor the FDA’s public dashboards quarterly to stay aware of common observation categories in your product class.
Most Commonly Cited Form 483 Violations
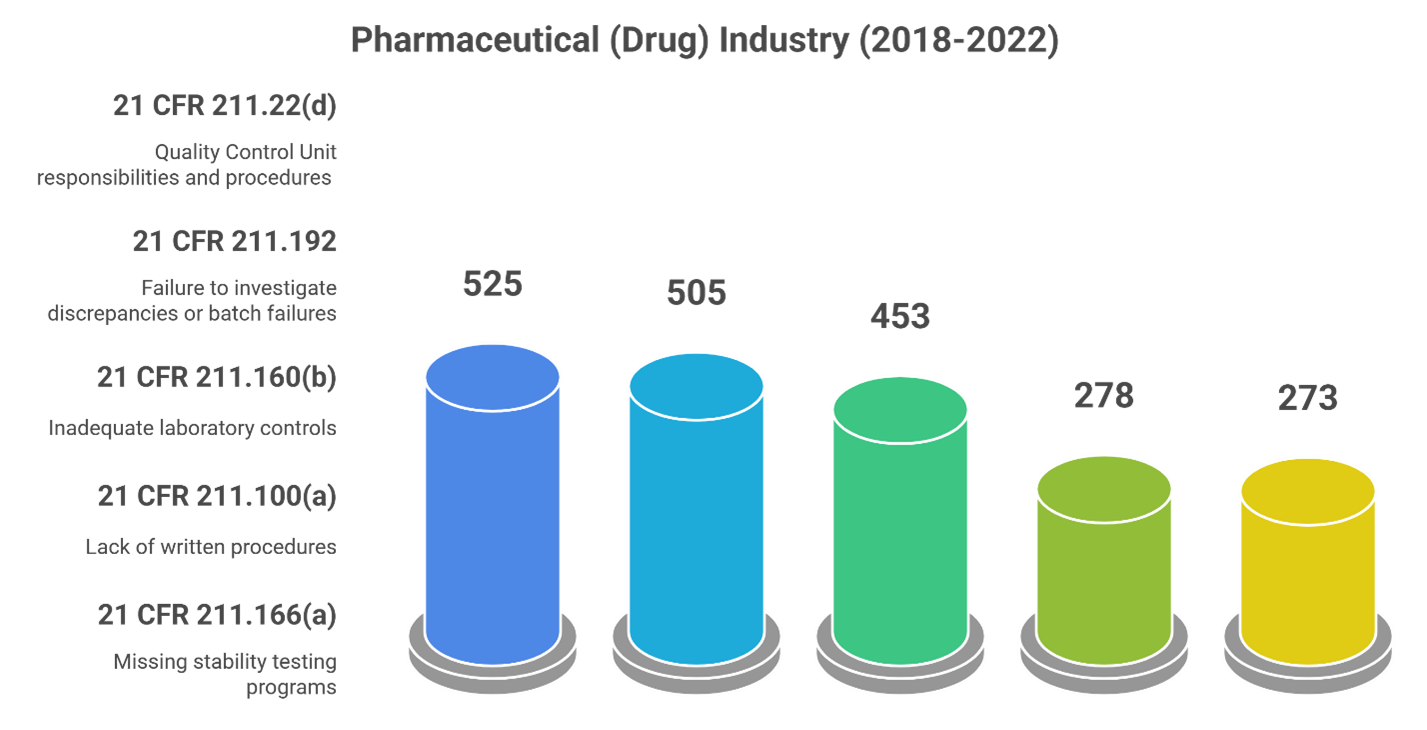
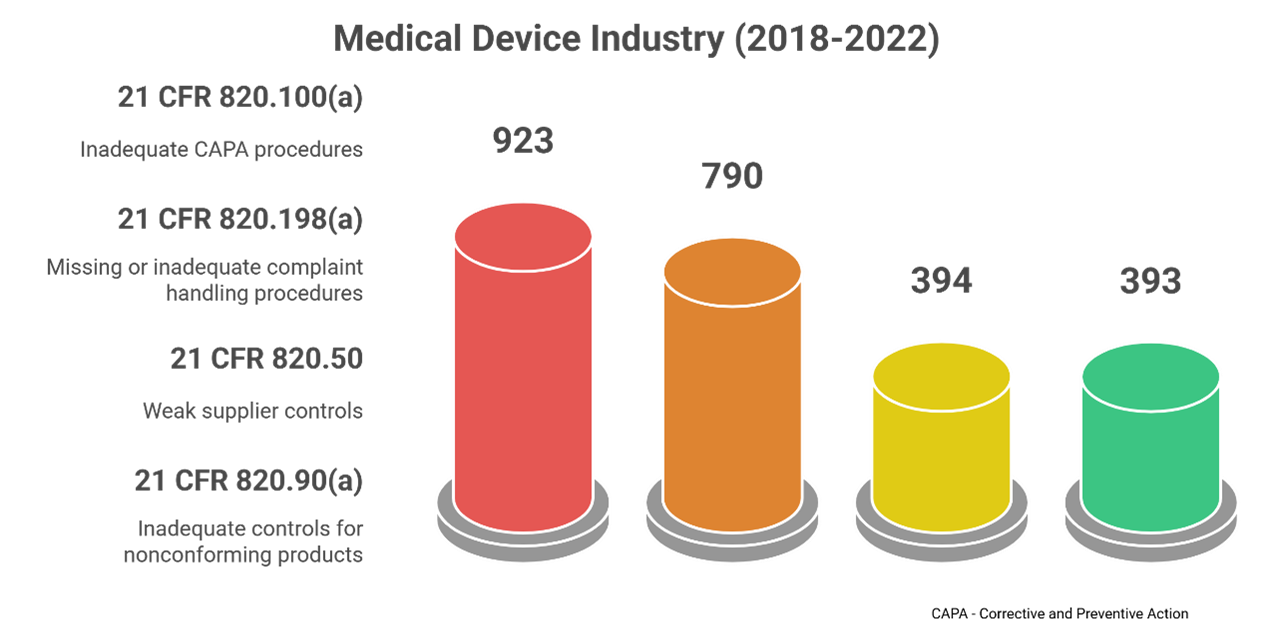
How Can Companies Prepare and Handle FDA Form 483?
Preventive Strategies: Inspection Readiness & Avoidance
- Conduct mock audits and gap assessments with a focus on cGMP, clinical trial compliance, eCTD documentation, and data integrity systems.
- Implement ongoing quality systems: SOP upkeep, training refreshers, automated audit trails, environmental monitoring plans, and periodic internal reviews.
- Foster a compliance culture: leadership buy-in, clear communication, and accountability across functional teams.
Best Practices: Swift, Strategic Response
- Internal review immediately: Discuss observations with the inspection team before they leave; clarify any misunderstandings and document discussions.
- Structured, timely written response: Submit within the 15‑business‑day window. Address each 483 item individually, including root‑cause analysis, corrective and preventive actions (CAPA), implementation timelines, and responsibilities. Use clear, evidence-based plans.
- Provide documentation: Corrected or updated SOPs, training logs, validation records, audit evidence, and timelines to FDA support credibility.
- Track execution: Monitor CAPA progress and evidence follow-through at scheduled intervals.
A well-organized, transparent, and timely response significantly reduces the risk of escalation to a Warning Letter or other enforcement action.
Tips at a Glance: Ensuring FDA Compliance
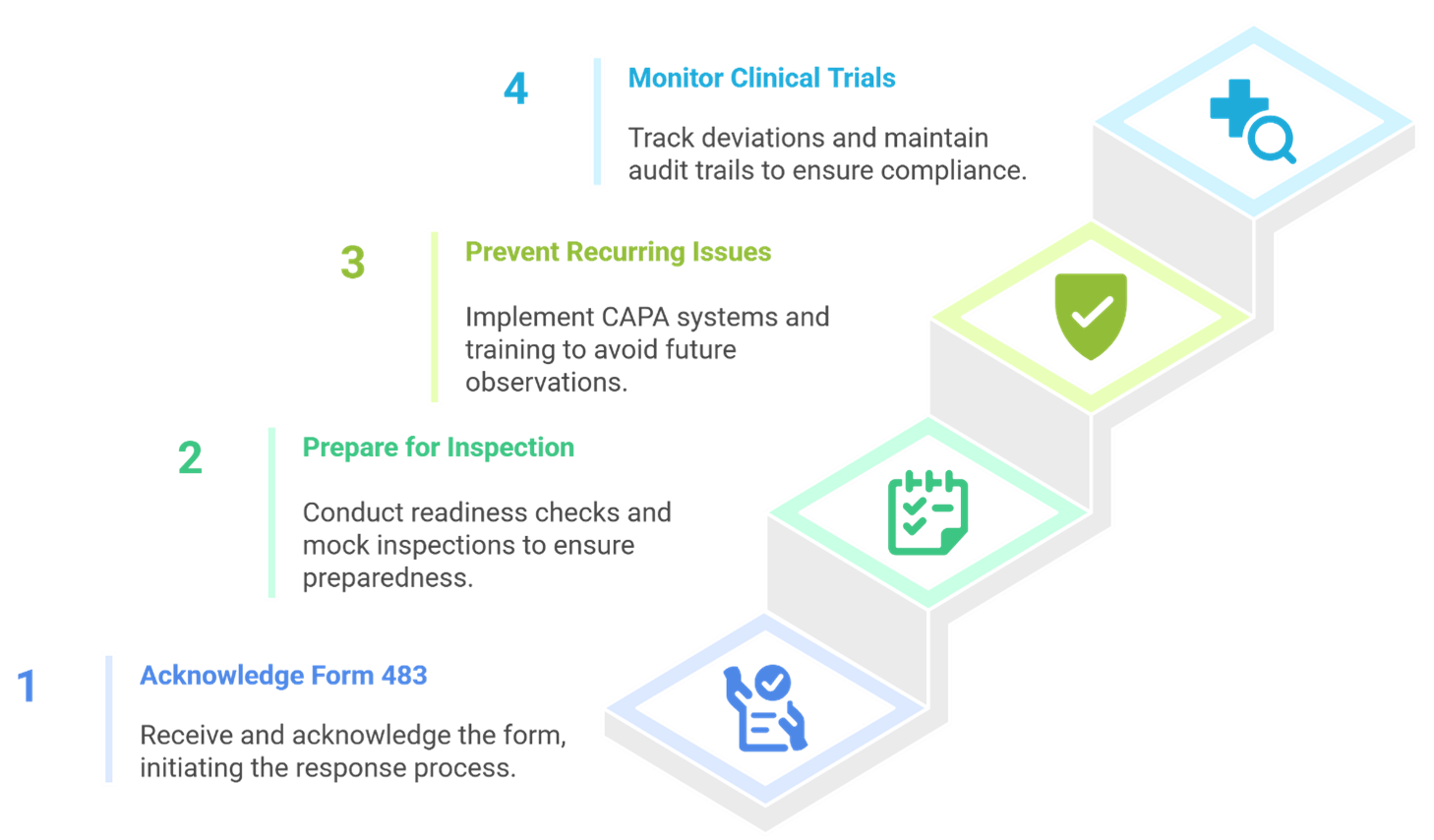
Closing Thoughts
An FDA Form 483 is a critical—but manageable—checkpoint in the regulatory lifecycle.
Proper preparation, rigorous systems, and expert support can turn inspection observations into opportunities for strengthening your compliance posture.
At BLA Regulatory, we partner with regulated organizations worldwide to anticipate risk, prepare teams, and respond with confidence. Whether you’re gearing up for a BLA or NDA inspection, analyzing protocol deviations, or updating GMP procedures, our services and expertise are designed for seamless alignment with FDA expectations.
Need help preparing for inspection, writing a response, or building CAPA systems? Reach out to BLA Regulatory for personalized assistance.